Fitting a Lennard-Jones potential for bulk Cu
In this tutorial you will learn how to create a dataset of structures, calculate their energies, and use the FitLJ package to fit a Lennard-Jones potential.
Creating the database
A necessary requirements for fitting an interatomic potential is to have a database of structures representative of the possible atomic configurations of the material of interest. The energies of these configurations are used as ground truth for the fitting procedure.
Here, we will use ASE to create a data set of different bulk structures of Cu at different volumes. To make the potenital as general and transferable as possible among different configuratiosn, we will consider three different crystalline systems:
Simple cubic:
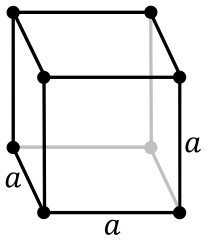
Face-centered cubic:
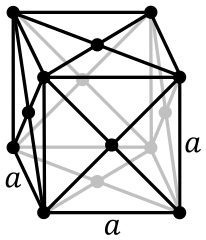
Body-centerd cubic:
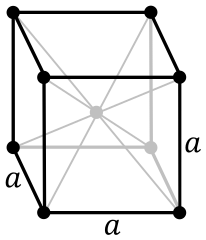
The volume of the three crystalline structures is defined by the lattice constant a. The ground truth energies will be calculated using the Effective Medium Theory.
Warning
The Effective Medium Theory used in this tutorial is not intended to produce accurate results and it is intended only for pedagogical purposes. For production simulations, one might want to use a more accurate and computationally intensive electronic structure method such as Density Functional Theory.
The script run_emt uses ASE to loop over different values of the lattice constant a for the three different crystalline structures. The calculated structures and associated energies are then dumped into a database. The latter will be used later for the fitting procedure.
1import numpy
2from ase import Atoms
3from ase.calculators.emt import EMT
4from ase.lattice.cubic import SimpleCubic, FaceCenteredCubic, BodyCenteredCubic
5from ase.db import connect
6
7# Define list of lattice constant values
8lattice_constants = numpy.linspace(2.0,6.0,10)
9
10# Connect to the database
11database = connect('Cu.db')
12
13# Loop over the crystalline lattice systems
14for lattice in [SimpleCubic, FaceCenteredCubic, BodyCenteredCubic]:
15
16 # Loop over the lattice constant values
17 for a in lattice_constants:
18
19 # Create the crystal unit cell
20 atoms = lattice('Cu',latticeconstant=a)
21
22 # Attach a calculator
23 atoms.calc = EMT()
24
25 # Get the potential energy
26 energy = atoms.get_potential_energy()
27
28 # Write the structure to the database
29 database.write(atoms)
Fitting the Lennard-Jones potential
Once the database has been created, the FitLJ program can be used to read the database and use it to fit the Lennard Jones potential parameters \(\sigma\) and \(\epsilon\).
The script fit load the database Cu.db previously created and fit the parameters of a Lennard Jones potenital to the energies of the structures contained in the database.
1from fitlj.fit_model import FitModel
2from fitlj.io import AtomicSimulationEnvironmentDataBase
3
4# Load the database file
5database = AtomicSimulationEnvironmentDataBase(database_filename='Cu.db')
6
7# Connect to the database
8database = database.connect_to_database()
9
10# Initial guess for sigma
11sigma_initial_guess = 15.0
12
13# Initial guess for epsilon
14epsilon_initial_guess = 3.5
15
16# Fit a Lennard Jones model to the configurations present in the database
17model = FitModel(database=database,
18 lennard_jones_parameters=[sigma_initial_guess, epsilon_initial_guess])
19sigma, epsilon = model.fit_lennard_jones_model()
20
21# Print the fitted values
22print(f'The fitted value of sigma is: {sigma:.5f} eV.')
23print(f'The fitted value of epsilon is: {epsilon:.5f} Ang.')
The optimized values of \(\sigma\) and \(\epsilon\) are printed after the optimization has successfully terminated:
1>>>python3 fit.py
2Optimization terminated successfully.
3 Current function value: 730.106876
4 Iterations: 22
5 Function evaluations: 72
6The fitted value of sigma is: 19.05469 eV.
7The fitted value of epsilon is: -0.50313 Ang.
Warning
In order for the optimization procedure to be succesfull, reasonable starting guesses for the \(\sigma\) and \(\epsilon\) parameters must be chosen. If the starting guesses are very far from the final, optimized values, the fitting procedure might not be successful.